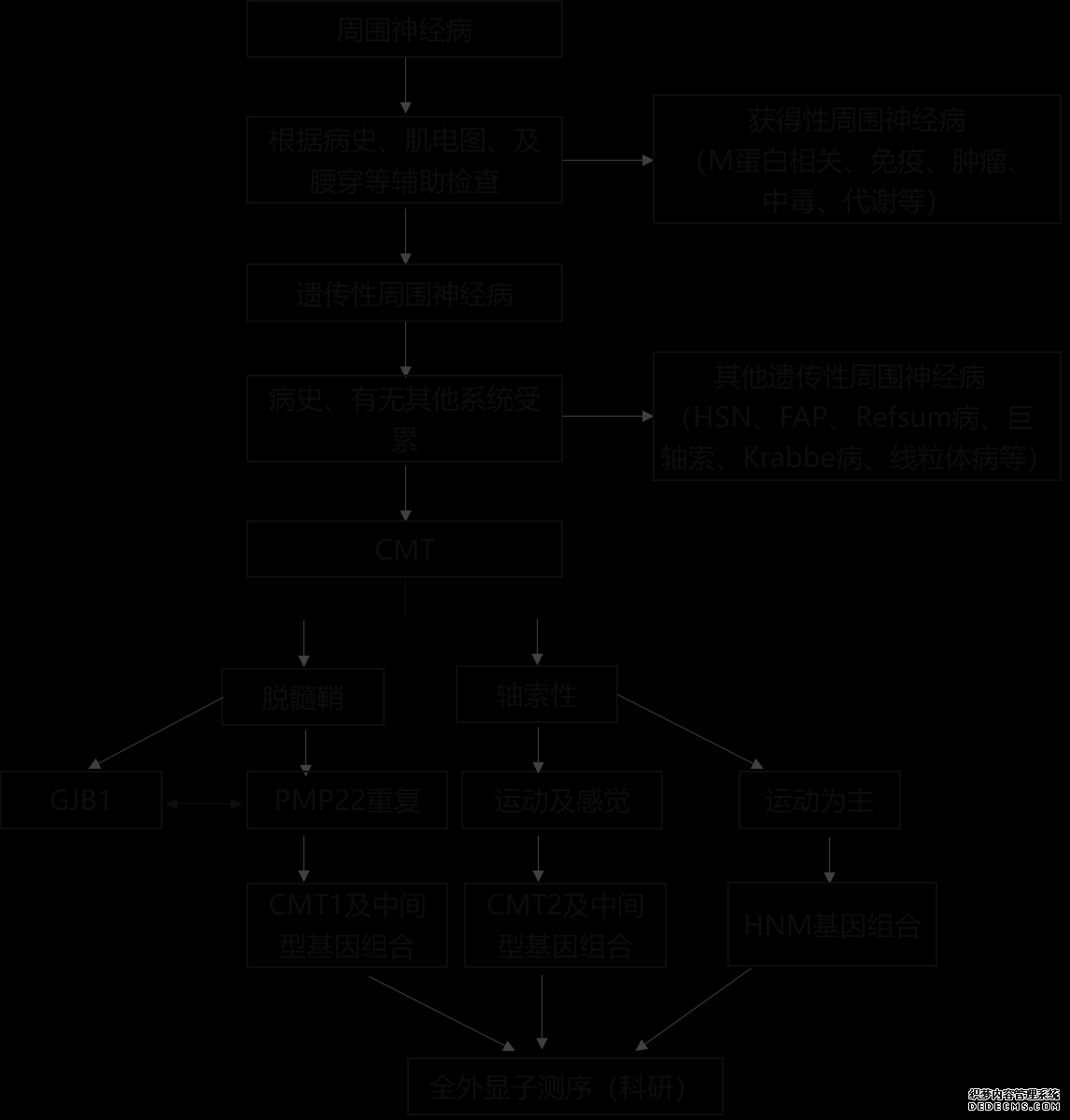
mt4交易软件官网肌电图及必要的肌肉活检病理及基因检测有助于鉴别诊断腓骨肌萎缩症(Charcot-Marie-Tooth disease,CMT)是一组遗传性方圆精神病。目前已发觉的致病基因达60余种。其重要特性为慢性实行性、长度依赖的运动及感触精神病,最常睹出现为下肢起病的、从容开展的肢体远端肌肉萎缩,无力和感触缺失。依据上肢运动神经传导速率重要分为髓鞘型和轴索型。依据遗传式样、临床出现以及电心理,CMT重要亚型征求CMT1-4以及CMTX。别的又有CMT5-7、dHMN(远端型遗传性运动精神病)、HNPP(遗传压迫易感方圆精神病)。正在每个亚型中,区别字母代外区别基因突变(如CMT1A,CMT1B)。
病因:CMT为一组由区别基因突变导致的方圆精神病。这些基因编码的卵白外达于方圆神经的髓鞘或轴索,突变导致方圆神经髓鞘造成缺陷或轴索功用卓殊。
遗传式样分为常染色体显性遗传、常染色体隐性遗传和X连锁隐性遗传等。常睹的突变基因征求PMP22,MPZ,GJB1,MFN2。常睹的亚型为CMT1,CMT2,CMTX。CMT1为常染色体显性遗传的脱髓鞘性CMT,此中CMT1A为最常睹的CMT亚型(占40~50%),其突变基由于PMP22;CMT1B占CMT1的3~5%,其突变基由于MPZ。X连锁隐性遗传的CMTX1为第2常睹的CMT亚型(占10%),其突变基由于GJB1。CMT2为轴利落CMT,此中常睹的突变基因征求MFN2(占CMT2的20%)、MPZ(占CMT2的5%)、NEFL和GDAP1等。CMT4为常染色体隐性遗传的脱髓鞘性CMT,其最常睹的突变基由于GDAP1。PMP22反复、GJB1突变、PMP22缺失、MPZ突变、MFN2突变这5种亚型占全盘CMT的92%。
盛行病学:CMT的总体发病率约为40/100 000,发病率正在人种间无明明不同。
临床出现:CMT的重要临床出现为下肢远端为主,并渐渐向近端繁荣的肢体肌肉萎缩、无力及感触丢失。常睹临床出现为运动才智不坊镳龄人,跑步贫穷,易扭脚,足下垂,小腿腓肠肌萎缩形似“鹤腿”;查体可发觉弓形足、锤状趾,远端肢体为主的无力萎缩,深感触减退。患者凡是20岁前起病,从容开展,疾病后期或者要紧影响举止,但很少导致全部残疾,也不影响平常寿命。但有些特地类型或者起病早且要紧:如Dejerine-Sottas归纳征患者婴儿期起病,导致低张力的软婴、运动发育迟滞等。少数CMT可有方圆精神病以外的其他出现:CMTX1型可有卒中样产生伴MRI白质可逆性病变;CMT5型伴锥体束征;CMT6型伴视神经萎缩;CMT7型伴色素性视网膜炎。
电心理查抄对待分别脱髓鞘性和轴利落精神病很是要紧,同时可能检测是否有临床下的感触神经受累,有助于CMT的分型;别的,节段性运动神经传导检测正在脱髓鞘型CMT与CIDP的甄别中也有要紧效用。匀称的神经传导速率减慢(上肢运动神经传导速率<38m/s)提示脱髓鞘型CMT(CMT1以及CMT4),而神经传导速率平常或轻度减慢(正中或尺神经运动传导速率>38m/s)、伴有复合肌肉行为电位及感触行为电位波幅低重提示CMT2。当上肢的运动神经传导速率位于25~45m/s的中央值时,必要警卫CMTX1。脱髓鞘型CMT的运动神经传导速率凡是匀称减慢,若浮现明明的波形离散、传导阻滞凡是提示CIDP或者性大。但正在MPZ基因突变的CMT1B中,偶然会浮现传导阻滞;CMTX1中,有时可有过错称的传导速率减慢,可有明明的波形离散乃至传导阻滞。
基因检测对待CMT的诊断和分型很是要紧。鉴于PMP22、MPZ、GJB1及MFN2基因突变正在CMT中占90%以上,故可能依据患者的临床和电心理特点选取或者相干的基因实行一代测序检测。跟着高通量测序身手的普及,对待常染色体显性的脱髓鞘性方圆精神病,可起首采用MLPA身手实行PMP22基因反复突变的检测,假设阴性再选取高通量测序伎俩对更众相干基因实行检测。
跟着基因检测伎俩运用,绝大大都疑诊病例无需实行神经活检。但当临床及肌电图不模范时,可通过神经活检来协助甄别诊断。
诊断:CMT的诊断仰仗临床出现和体格查抄、电心理查抄及基因检测。对待从容开展的肢体远端肌肉无力萎缩、弓形足、伴或不伴有轻度感触卓殊,电心理提示感触运动性方圆精神病的患者,无论有无阳性家族史,需商讨到遗传性方圆精神病,迥殊是CMT。基因检测是确诊CMT及实行分型的中央本领。
产前诊断:对待要紧致残的类型,正在家眷弥漫知情、包罗看法后,可商讨再次生育时实行产前诊断。
1.CMT重要必要与极少累及方圆神经的其他遗传性疾病相甄别,如Krabbe脑白质养分不良、异染性脑白质养分不良、线粒体病、遗传性痉挛性截瘫和遗传性共济失调等。它们除具有方圆精神病以外,又有神经编制其他部位和非神经结构器官受累的出现,而CMT较少有方圆神经以外的其他编制受累。另极少以方圆神经受累为主的遗传性病,如远端遗传性运动精神病(dHMN)、Refsum病、家族性淀粉样变性、巨轴索精神病和遗传性压迫易感方圆精神病等,必要正在临床和电心理查抄根底上,选取须要的生化检修、神经活检病理和基因检测来加以甄别。别的,还必要与远端型肌病和下运动神经元归纳征(如脊肌萎缩症)相甄别,肌电图及须要的肌肉活检病理及基因检测有助于甄别诊断。
2.CMT必要与得回性方圆精神病相甄别,如CIDP(慢性炎性脱髓鞘性众发神经根方圆精神病)、副卵白血症相干方圆精神病,轴利落如中毒、代谢相干方圆精神病和众灶运动精神病等。CMT凡是正在青少年或年少起病,起病年事晚需警卫得回性方圆精神病。CMT众起病藏匿、数年内从容加重,而得回性方圆精神病众病程较短。查体发觉弓形足、锤状趾、鹤腿症状提示CMT或者性大。脱髓鞘型CMT的运动神经传导速率凡是匀称减慢,若浮现明明的波形离散、传导阻滞,凡是提示CIDP或者性大。CMT的脑脊液卵白可轻度升高,但若明明升高(如>1g/L)则需商讨CIDP等得回性方圆精神病或者。
疗养:目前,CMT的疗养重要是接济疗养,没有刷新疾病的特异性药物。合意的接济疗养也许明显刷新患者的存在质料。
典范的痊愈疗养也许延缓疾病形成的功用袭击如合节异常等,维护更好的存在功用和容貌。支具鞋等可刷新行走步态。
如长春新碱、胺碘酮、硼替佐米、铂类、氨苯砜、来氟米特、呋喃妥因、甲硝唑、司他夫定、他克莫司、沙利度胺、扎西他滨等。
CMT类型繁众,基因确诊后提倡遗传商讨,明了病因及家系成员危机。对待要紧致残的类型,正在家眷弥漫知情、包罗看法后,可商讨再次生育时实行产前诊断。
遗传商讨:CMT类型繁众,基因确诊后提倡遗传商讨,明了病因及家系成员危机。
遗传式样分为常染色体显性遗传、常染色体隐性遗传和X连锁隐性遗传等。常睹的突变基因征求PMP22,MPZ,GJB1,MFN2。常睹的亚型为CMT1,CMT2,CMTX。CMT1为常染色体显性遗传的脱髓鞘性CMT,此中CMT1A为最常睹的CMT亚型(占40~50%),其突变基由于PMP22;CMT1B占CMT1的3~5%,其突变基由于MPZ。X连锁隐性遗传的CMTX1为第2常睹的CMT亚型(占10%),其突变基由于GJB1。CMT2为轴利落CMT,此中常睹的突变基因征求MFN2(占CMT2的20%)、MPZ(占CMT2的5%)、NEFL和GDAP1等。CMT4为常染色体隐性遗传的脱髓鞘性CMT,其最常睹的突变基由于GDAP1。PMP22反复、GJB1突变、PMP22缺失、MPZ突变、MFN2突变这5种亚型占全盘CMT的92%。
预后:因病程开展从容,预后尚好。疾病后期或者要紧影响举止,但很少导致全部残疾,也不影响平常寿命。
英文名:Charcot-Marie-Tooth disease 简称:CMT 别称:遗传性运动感触精神病 科室:神经内科 症状:下肢肌肉萎缩、运动底劣等;
转载请注明出处。

 相关文章
相关文章


 精彩导读
精彩导读




 热门资讯
热门资讯 关注我们
关注我们
